Dendrograma de agrupamento hierárquico com suporte bootstrap [Portuguese text]
Aprenda a gerar um dendrograma de agrupamento hierárquico com suporte bootstrap.
Neste tutorial você aprenderá a gerar um dendrograma de Agrupamento Hierárquico (Hierarchical Cluster Analysis) usando a medida de dissimilaridade de Gower (Gower 1971) e o método de agrupamento Ward.D2 (Murtagh and Legendre 2014), além de adicionar valores de Bootstrap (Efron, Halloran, and Holmes 1996; Felsenstein 1985) aos nós do dendrograma para avaliar a incerteza na análise de agrupamento. A ideia do post não é entrar no mérito de como cada método funciona, mas sim em mostrar como isso pode ser implementado no R.
Esta postagem foi inteiramente produzida em ambiente R Studio, usando os pacotes rmarkdown (Allaire et al. 2021; Xie, Allaire, and Grolemund 2018; Xie, Dervieux, and Riederer 2020), rmdformats (Barnier 2021), remedy (Fay, Sidi, and Smith 2018), knitr (Xie 2014, 2015, 2021) e citr (Aust 2019).
1. Instalação dos pacotes necessários
Antes de iniciarmos, é importante que você tenha os seguintes pacotes e dependências instalados:
downloader(Chang 2015)parallel(R Core Team 2020)cluster(Maechler et al. 2019)dendextend(Galili 2015)
#instala os pacotes, se for necessário:
pacotes = c("downloader","parallel","cluster","dendextend")
for (p in setdiff(pacotes, installed.packages()[,"Package"])) {install.packages(p, dependencies=TRUE)}Para gerar os valores bootstrap você precisará executar os comandos a seguir para baixar o arquivo contendo as funções necessárias. Originalmente, essas funções foram implementadas no pacote booststrap por Sebastian Gibb (2013) e adaptadas para este tutorial.
#faz o download das funções necessárias para o seu diretório de trabalho a partir do meu repositório no Github
downloader::download("https://github.com/CarolineVasconcelos/FunctionsFromBootstrapPackage/archive/refs/heads/main.zip",
dest="FunctionsFromBootstrapPackage-main.zip",
mode="wb")
#extrai o arquivo
utils::unzip("FunctionsFromBootstrapPackage-main.zip", exdir=".")
#carrega o arquivo contendo as funções necessárias
source("FunctionsFromBootstrapPackage-main/funcoesbootstrap.R")2. Prepara os dados
Aqui, vamos usar como exemplo o Iris Dataset (Anderson 1935), que consiste em um data.frame com 150 observações (linhas) e 5 variáveis (colunas). Este famoso conjunto de dados fornece as medidas em centímetros das variáveis denominadas Sepal.Length, Sepal.Width, Petal.Length, Petal.Width e Species, respectivamente, para 50 flores de cada espécie de Iris (I. setosa, I. versicolor e I. virginica).
data(iris) #exemplo com o iris dataset
head(iris) #checa o topo dos dados## Sepal.Length Sepal.Width Petal.Length Petal.Width Species
## 1 5.1 3.5 1.4 0.2 setosa
## 2 4.9 3.0 1.4 0.2 setosa
## 3 4.7 3.2 1.3 0.2 setosa
## 4 4.6 3.1 1.5 0.2 setosa
## 5 5.0 3.6 1.4 0.2 setosa
## 6 5.4 3.9 1.7 0.4 setosastr(iris) #checa a estrutura dos dados## 'data.frame': 150 obs. of 5 variables:
## $ Sepal.Length: num 5.1 4.9 4.7 4.6 5 5.4 4.6 5 4.4 4.9 ...
## $ Sepal.Width : num 3.5 3 3.2 3.1 3.6 3.9 3.4 3.4 2.9 3.1 ...
## $ Petal.Length: num 1.4 1.4 1.3 1.5 1.4 1.7 1.4 1.5 1.4 1.5 ...
## $ Petal.Width : num 0.2 0.2 0.2 0.2 0.2 0.4 0.3 0.2 0.2 0.1 ...
## $ Species : Factor w/ 3 levels "setosa","versicolor",..: 1 1 1 1 1 1 1 1 1 1 ...summary(iris) #faz um resumo dos dados## Sepal.Length Sepal.Width Petal.Length Petal.Width
## Min. :4.300 Min. :2.000 Min. :1.000 Min. :0.100
## 1st Qu.:5.100 1st Qu.:2.800 1st Qu.:1.600 1st Qu.:0.300
## Median :5.800 Median :3.000 Median :4.350 Median :1.300
## Mean :5.843 Mean :3.057 Mean :3.758 Mean :1.199
## 3rd Qu.:6.400 3rd Qu.:3.300 3rd Qu.:5.100 3rd Qu.:1.800
## Max. :7.900 Max. :4.400 Max. :6.900 Max. :2.500
## Species
## setosa :50
## versicolor:50
## virginica :50
##
##
## #cria um identificador ÚNICO para as amostras
#Por exemplo, pode ser o voucher ou qualquer outro código
minhasAmostras = paste(iris[,5],"-", seq(1:length(iris[,5])), sep="")
#junta esse identificador aos dados originais
meusDados = data.frame(iris, minhasAmostras)
#nomeia as linhas do data.frame a partir dos identificadores
rownames(meusDados) = minhasAmostras
#mostra isso
head(meusDados)## Sepal.Length Sepal.Width Petal.Length Petal.Width Species
## setosa-1 5.1 3.5 1.4 0.2 setosa
## setosa-2 4.9 3.0 1.4 0.2 setosa
## setosa-3 4.7 3.2 1.3 0.2 setosa
## setosa-4 4.6 3.1 1.5 0.2 setosa
## setosa-5 5.0 3.6 1.4 0.2 setosa
## setosa-6 5.4 3.9 1.7 0.4 setosa
## minhasAmostras
## setosa-1 setosa-1
## setosa-2 setosa-2
## setosa-3 setosa-3
## setosa-4 setosa-4
## setosa-5 setosa-5
## setosa-6 setosa-6#cria um objeto contendo apenas as variáveis
minhasVariaveis = meusDados[,-c(5:6)]
#mostra isso
head(minhasVariaveis)## Sepal.Length Sepal.Width Petal.Length Petal.Width
## setosa-1 5.1 3.5 1.4 0.2
## setosa-2 4.9 3.0 1.4 0.2
## setosa-3 4.7 3.2 1.3 0.2
## setosa-4 4.6 3.1 1.5 0.2
## setosa-5 5.0 3.6 1.4 0.2
## setosa-6 5.4 3.9 1.7 0.43. Bootstrapping simples para agrupamento hierárquico
Agora vamos gerar os valores bootstrap e plotá-los no dendrograma:
#função que cria um objeto 'hclust' pelo método Ward.D2 usando uma matriz de distância pelo método de Gower.
#IMPORTANTE: No argumento "type" é importante especificar o tipo de cada variável em uma lista [list()].
#No caso do 'iris dataset' todas as variáveis são númericas. Veja também o help da função daisy()
createHclustObject = function(data) hclust(cluster::daisy(data,
metric="gower", #dissimilaridade de Gower
type=list(numeric=c(1:4))), #especifica variáveis
method="ward.D2") #método de agrupamento
require("parallel")
resultadoBootstrap = bootstrap(minhasVariaveis,
fun=createHclustObject,
n=1000L) #faz booststrap para n = 1000 vezes
hclust = createHclustObject(minhasVariaveis) #gera um objeto da classe hclust
meuDendrograma = as.dendrogram(hclust) #converte o hclust em um dendrograma
#define alguns parâmetros gráficos
par(mar=c(7,5,4,1))
#plota o dendrograma
plot(meuDendrograma,
ylab="Dissimilaridade de Gower", cex.lab=1.5, #eixo Y
main="Dendrograma de Agrupamento Hierárquico", sub="Método de agrupamento Ward.D2", cex.main=1.8, #título
nodePar=list(lab.cex=1, pch=NA), #define alguns parâmetros dos nós
type="rectangle") #ou pode usar também type="triangle"
#plota os valores de booststrap correspondentes a cada nó do dendrograma
bootlabels.hclust(hclust, resultadoBootstrap, col="blue", cex=0.9)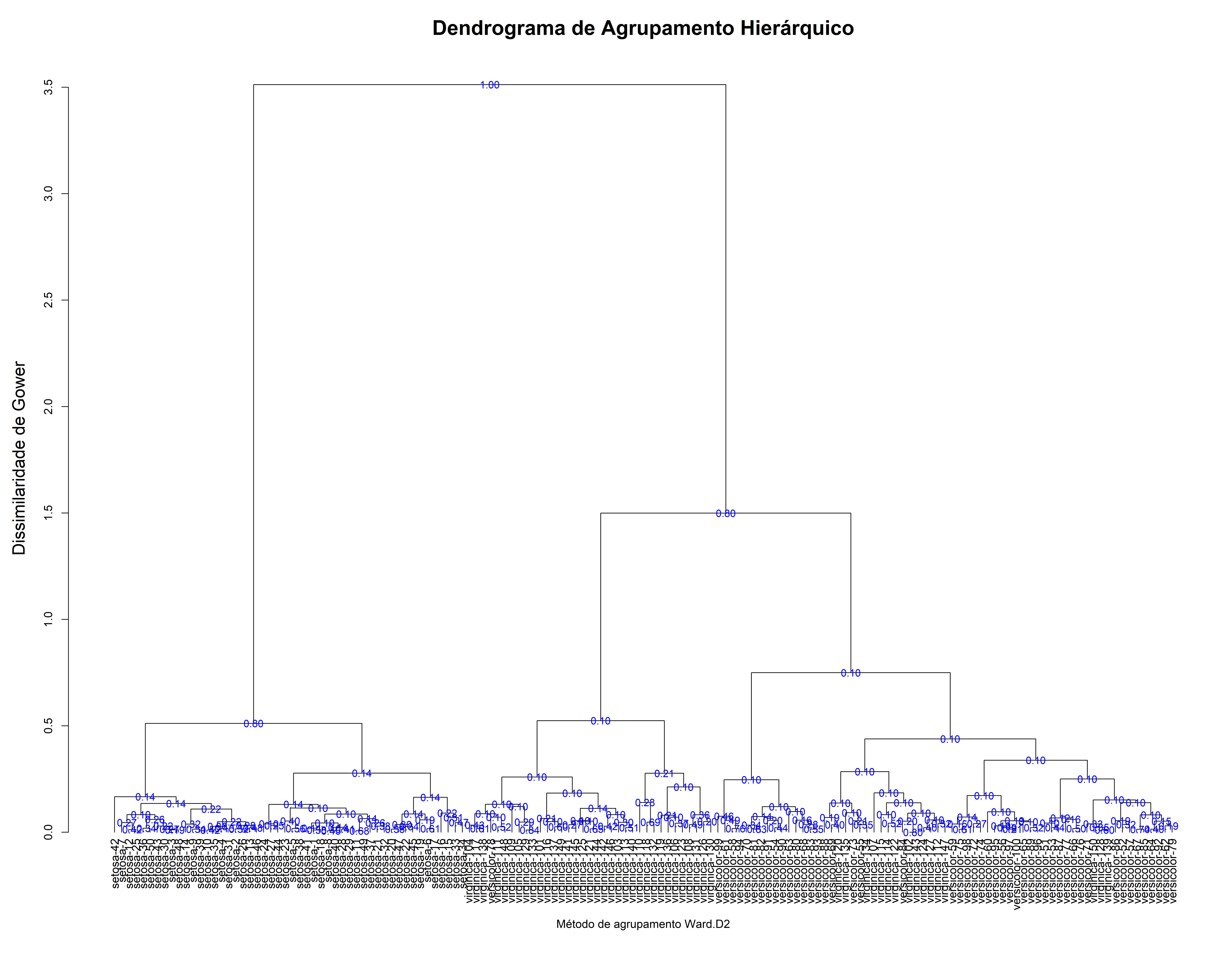
4. Melhorando a visualização do dendrograma
Opção 1 - Dendrograma com barras coloridas indicando categorias (spp) e grupos (clusters)
#quais amostras formam k grupos?
grupos = cutree(hclust, k=3) #define um k
#inclui no data.frame original uma coluna contendo o nome do grupo ao qual cada amostra faz parte
meusDados$clusterID = grupos
#checa o resultado disso
head(meusDados)## Sepal.Length Sepal.Width Petal.Length Petal.Width Species
## setosa-1 5.1 3.5 1.4 0.2 setosa
## setosa-2 4.9 3.0 1.4 0.2 setosa
## setosa-3 4.7 3.2 1.3 0.2 setosa
## setosa-4 4.6 3.1 1.5 0.2 setosa
## setosa-5 5.0 3.6 1.4 0.2 setosa
## setosa-6 5.4 3.9 1.7 0.4 setosa
## minhasAmostras clusterID
## setosa-1 setosa-1 1
## setosa-2 setosa-2 1
## setosa-3 setosa-3 1
## setosa-4 setosa-4 1
## setosa-5 setosa-5 1
## setosa-6 setosa-6 1#cria um objeto para armazenar cores
#você pode incluir mais cores ou usar uma paleta pronta
#conforme o número de grupos que quer testar
coresCategorias = c("#CC476B", #rosa
"#309210", #azul
"#0082CE") #verde
#vamos criar barras para plotar junto ao dendrograma
#aqui as barras são configuradas conforme as categorias (spp) e grupos (clusters)
#atribui cores às "Species" na mesma ordem do dendrograma
barraSpp = coresCategorias[meusDados$Species][order.dendrogram(meuDendrograma)]
#atribui cores aos grupos "clusterID"
barraGrp = coresCategorias[meusDados$clusterID]
#une as barras
barras = cbind(barraSpp, barraGrp)
#define alguns parâmetros gráficos - pois a figura pode ficar grande demais
par(mar=c(10,5,4,1) + 0.1, xpd=NA)
#define alguns parâmetros em relação aos nós do dendrograma
nP = list(col=c(NA, NA),
cex=c(3, 0.5),
pch=c(21, NA),
bg=c("gray66", "black"),
lab.cex=1,
lab.col="black")
#plota o dendrograma
plot(meuDendrograma,
nodePar=nP,
edgePar=list(col="gray66", lwd=1.2), #define alguns parâmetros dos ramos
horiz=FALSE, #se horiz=TRUE a plotagem dos valores bootstraps fica prejudicada
ylab="Dissimilaridade de Gower", cex.lab=1.5, #eixo Y
main="Dendrograma de Agrupamento Hierárquico", sub="Método de agrupamento Ward.D2", cex.main=1.8) #título
#plota os valores de bootstrap
bootlabels.hclust(hclust, resultadoBootstrap, col="darkblue", cex=0.7)
library(dendextend) #carrega o pacote que contém a função colored_bars()
#plota as barras
colored_bars(colors=barras, dend=meuDendrograma, rowLabels=c("Espécies","Grupos"))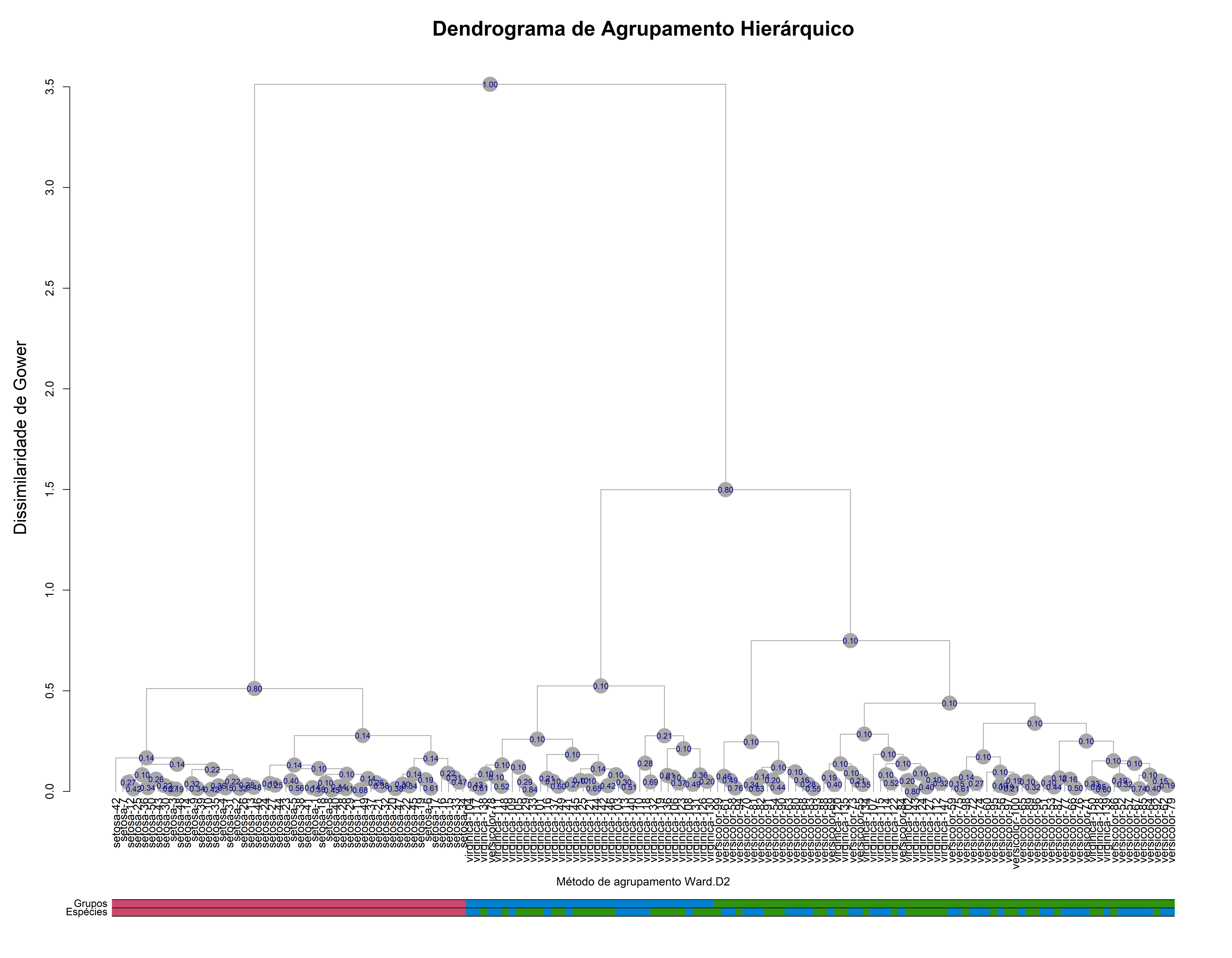
Opção 2 - Dendrograma com labels coloridos em vez das barras
par(mar=c(10,5,4,1) + 0.1, xpd=NA) #define alguns parâmetros gráficos
library(dendextend) #carrega o pacote que contém a função labels_colors()
#para colorir os labels
labels_colors(meuDendrograma) = coresCategorias[meusDados$Species][order.dendrogram(meuDendrograma)]
#define alguns parâmetros em relação aos nós do dendrograma
nP = list(col=c(NA, NA),
cex=c(3, 0.5),
pch=c(21, NA),
bg=c("gray66", "black"),
lab.cex=1,
lab.col="black")
#plota o dendrograma
plot(meuDendrograma,
nodePar=nP,
edgePar=list(col="gray66", lwd=1.2), #define alguns parâmetros dos ramos
horiz=F, #se horiz=TRUE a plotagem dos valores bootstraps fica prejudicada
ylab="Distância de Gower", cex.lab=1.5, #eixo Y
main="Dendrograma de Agrupamento Hierárquico", sub="Método de agrupamento Ward.D2", cex.main=1.8) #título
#plota os valores de bootstrap
bootlabels.hclust(hclust, resultadoBootstrap, col="darkblue", cex=0.7)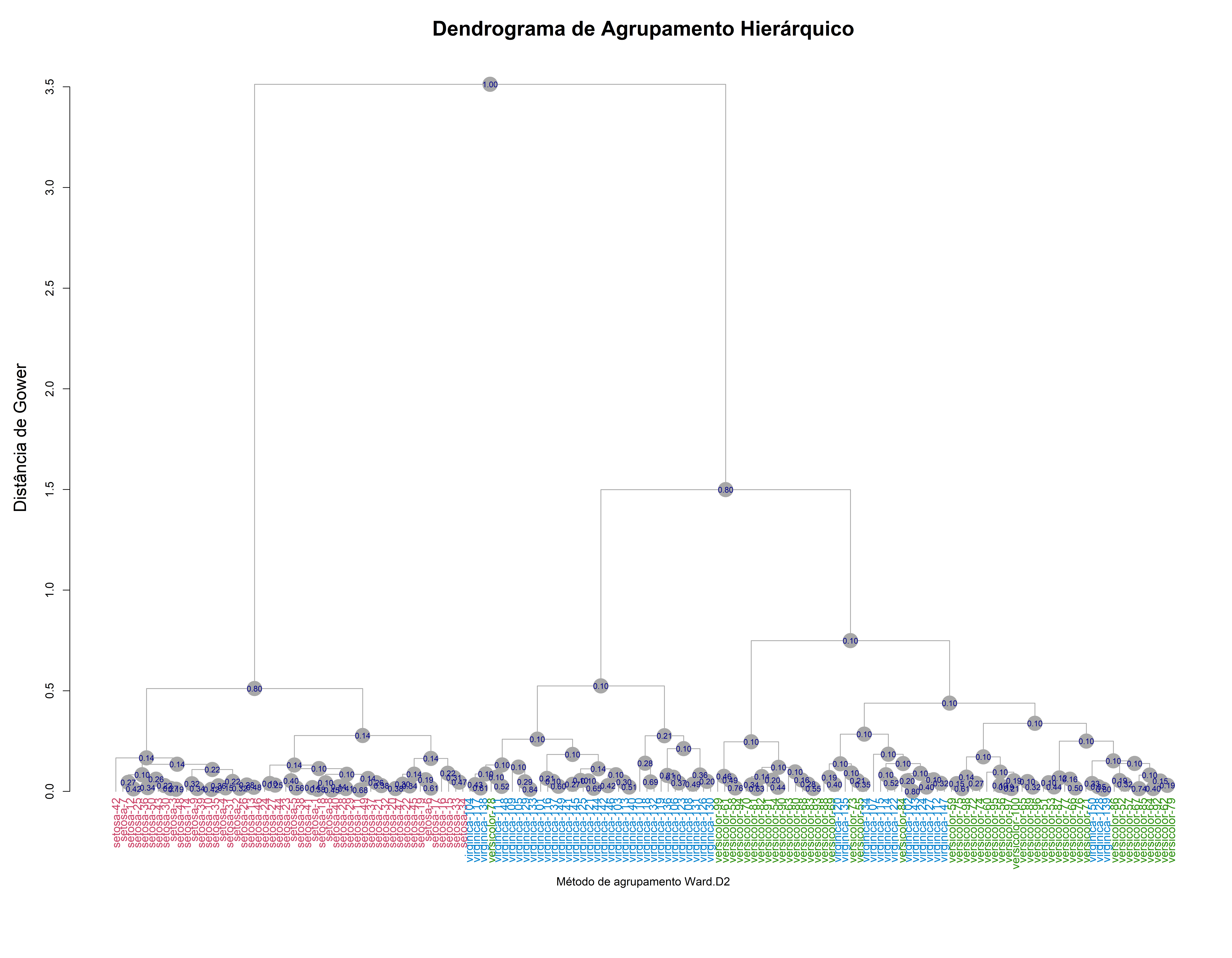
5. Salvando as figuras em PDF
Para salvar uma figura use as seguintes funções pdf() e dev.off(). Veja como no exemplo abaixo:
pdf("FigDendrograma_labelscoloridos.pdf", width=18, height=14) #abre um dispositivo pdf para salvar a figura
#plota o dendrograma
plot(meuDendrograma,
nodePar=nP,
edgePar=list(col="gray66", lwd=1.2), #define alguns parâmetros dos ramos
horiz=FALSE, #se horiz=TRUE a plotagem dos valores bootstraps fica prejudicada
ylab="Dissimilaridade de Gower", cex.lab=1.5, #eixo Y
main="Dendrograma de Agrupamento Hierárquico", sub="Método de agrupamento Ward.D2", cex.main=1.8) #título
#plota os valores de bootstrap
bootlabels.hclust(hclust, resultadoBootstrap, col="darkblue", cex=0.7)
dev.off() #fecha o dispositivo e salva a figura## png
## 2Postagem originalmente publicada no meu perfil do RPubs. Toda semana tem conteúdo novo por lá!
Até a próxima! See you soon!
Referências
Allaire, JJ, Yihui Xie, Jonathan McPherson, Javier Luraschi, Kevin Ushey, Aron Atkins, Hadley Wickham, Joe Cheng, Winston Chang, and Richard Iannone. 2021. Rmarkdown: Dynamic Documents for R. https://github.com/rstudio/rmarkdown.
Anderson, Edgar. 1935. “The Irises of the Gaspe Peninsula.” Bulletin of the American Iris Society 59: 2–5.
Aust, Frederik. 2019. Citr: ’RStudio’ Add-in to Insert Markdown Citations. https://github.com/crsh/citr.
Barnier, Julien. 2021. Rmdformats: HTML Output Formats and Templates for ’Rmarkdown’ Documents. https://CRAN.R-project.org/package=rmdformats.
Chang, Winston. 2015. Downloader: Download Files over Http and Https. https://CRAN.R-project.org/package=downloader.
Efron, Bradley, Elizabeth Halloran, and Susan Holmes. 1996. “Bootstrap Confidence Levels for Phylogenetic Trees.” Proceedings of the National Academy of Sciences 93 (23): 13429–9. https://doi.org/10.1073/pnas.93.23.13429.
Fay, Colin, Jonathan Sidi, and Luke Smith. 2018. Remedy: ’RStudio’ Addins to Simplify ’Markdown’ Writing. https://CRAN.R-project.org/package=remedy.
Felsenstein, Joseph. 1985. “Confidence Limits on Phylogenies: An Approach Using the Bootstrap.” Evolution 39 (4): 783–91. https://doi.org/10.1111/j.1558-5646.1985.tb00420.x.
Galili, Tal. 2015. “Dendextend: An R Package for Visualizing, Adjusting, and Comparing Trees of Hierarchical Clustering.” Bioinformatics. https://doi.org/10.1093/bioinformatics/btv428.
Gower, John C. 1971. “A General Coefficient of Similarity and Some of Its Properties.” Biometrics, 857–71. https://doi.org/10.2307/2528823.
Maechler, Martin, Peter Rousseeuw, Anja Struyf, Mia Hubert, and Kurt Hornik. 2019. Cluster: Cluster Analysis Basics and Extensions.
Murtagh, Fionn, and Pierre Legendre. 2014. “Ward’s Hierarchical Agglomerative Clustering Method: Which Algorithms Implement Ward’s Criterion?” Journal of Classification 31 (3): 274–95. https://doi.org/10.1007/s00357-014-9161-z.
R Core Team. 2020. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing. https://www.R-project.org/.
Xie, Yihui. 2014. “Knitr: A Comprehensive Tool for Reproducible Research in R.” In Implementing Reproducible Computational Research, edited by Victoria Stodden, Friedrich Leisch, and Roger D. Peng. Chapman; Hall/CRC. http://www.crcpress.com/product/isbn/9781466561595.
———. 2015. Dynamic Documents with R and Knitr. 2nd ed. Boca Raton, Florida: Chapman; Hall/CRC. https://yihui.org/knitr/.
———. 2021. Knitr: A General-Purpose Package for Dynamic Report Generation in R. https://yihui.org/knitr/.
Xie, Yihui, J. J. Allaire, and Garrett Grolemund. 2018. R Markdown: The Definitive Guide. Boca Raton, Florida: Chapman; Hall/CRC. https://bookdown.org/yihui/rmarkdown.
Xie, Yihui, Christophe Dervieux, and Emily Riederer. 2020. R Markdown Cookbook. Boca Raton, Florida: Chapman; Hall/CRC. https://bookdown.org/yihui/rmarkdown-cookbook.
Caroline C. Vasconcelos
Research Fellow
My research interests include taxonomy and systematics (especially neotropical Sapotaceae), spectroscopy as a integrative tools, Amazonian flora, species distribution modeling, floristic studies, and tropical forest ecology.